Interesting paper about what LC-MS mode you should pick for quantitative approach.
Abstract:
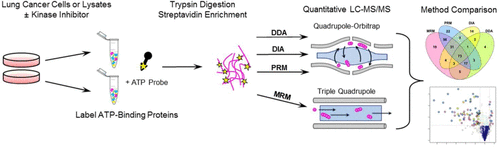
However, detection of kinases in biological matrices can be challenging; therefore, activity-based protein profiling enrichment of ATP-utilizing proteins was selected as a test case for exploring the limits of detection of low-abundance analytes in complex biological samples.
- To examine the impact of different MS acquisition platforms, quantification of kinase ATP uptake following kinase inhibitor treatment was analyzed by four different methods: LC–MS/MS with DDA and DIA, LC–MRM, and LC–PRM.
- For discovery data sets, DIA increased the number of identified kinases by 21% and reduced missingness when compared with DDA. In this context, MRM and PRM were most effective at identifying global kinome responses to inhibitor treatment, highlighting the value of a priori target identification and manual evaluation of quantitative proteomics data sets.
- We compare results for a selected set of desthiobiotinylated peptides from PRM, MRM, and DIA and identify considerations for selecting a quantification method and postprocessing steps that should be used for each data acquisition strategy.
Melissa A. Hoffman†‡, Bin Fang†, Eric B. Haura†, Uwe Rix†, and John M. Koomen*†
†H. Lee Moffitt Cancer Center & Research Institute, Tampa, Florida 33612-9497, United States
‡ Cancer Biology Ph.D. Program, University of South Florida, Tampa, Florida 33620, United States
J. Proteome Res.